Research
We develop the next generation of machine learning (ML) methods for addressing fundamental problems in biology.
Our major motivation is the rapid development of new biological technologies – including single-cell/spatial sequencing and CRISPR – which measure diverse molecular modalities at unprecedented throughput and resolution. ML methods are essential for analyzing and interpreting these large, high-dimensional, and multi-modal biological datasets. However, standard “off-the-shelf” ML methods are severely challenged by the high noise, sparsity, heterogeneity, and other limitations of modern biological technologies.
We address these challenges by developing ML methods that can extract meaningful biological insights from noisy biomedical data. Our research involes developing both new mathematical theory and practical algorithms. We draw on techniques from many different disciplines including deep learning, graph theory, statistical inference, and pure mathematics (e.g. complex analysis).
ML for spatial biology
|
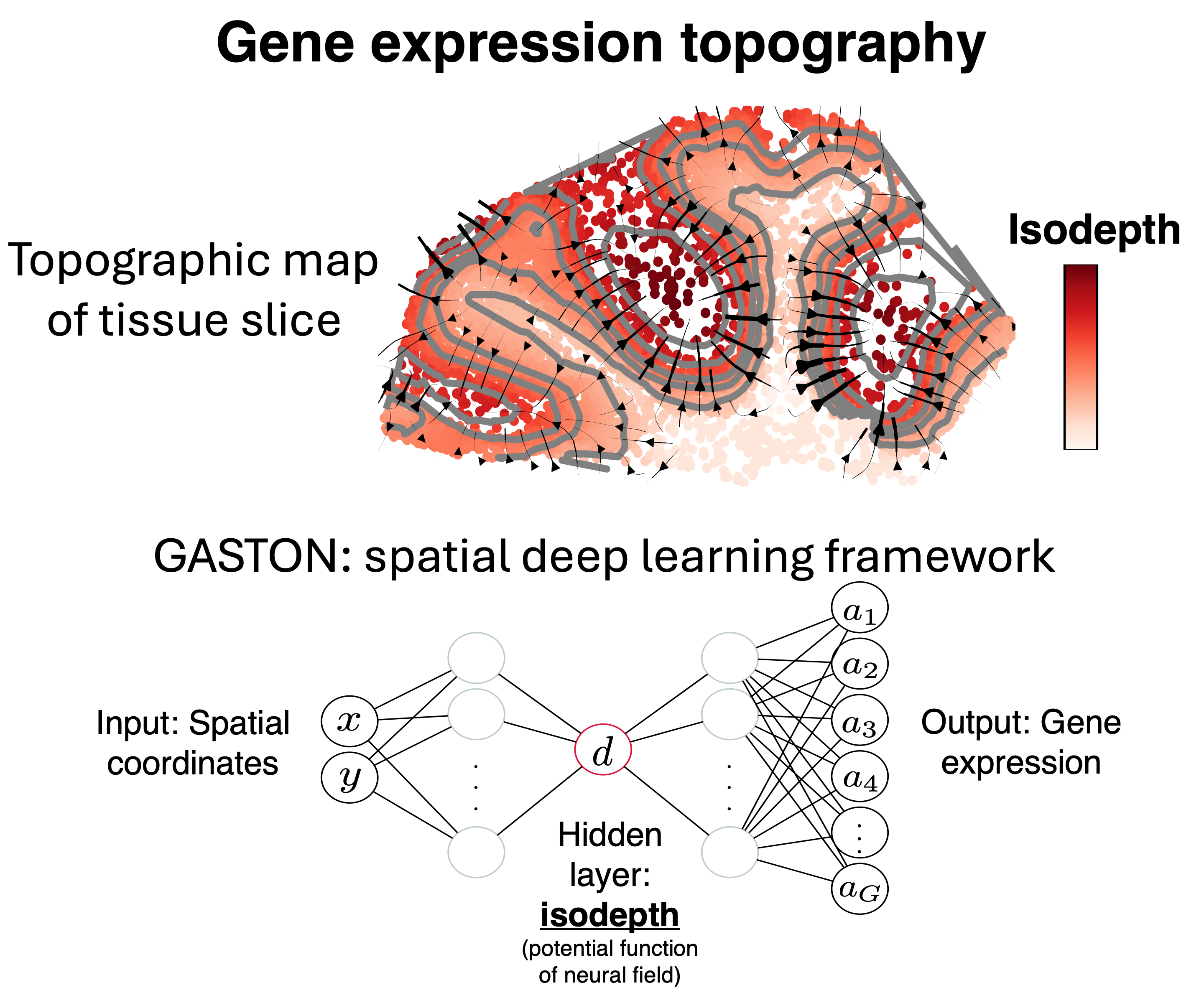
Next-generation spatial sequencing technologies measures both high-throughput cellular measurements (e.g. gene expression) and the spatial location of measured cells. We develop spatial ML methodologies that model the latent geometric structure of biological tissues from sparse spatial sequencing data. Our goal is to quantify cellular dynamics over space and time. Key words: spatial transcriptomics, cell-cell interactions, spatial gradients, deep learning, copula, time-series analysis
|
|
Biological networks
|
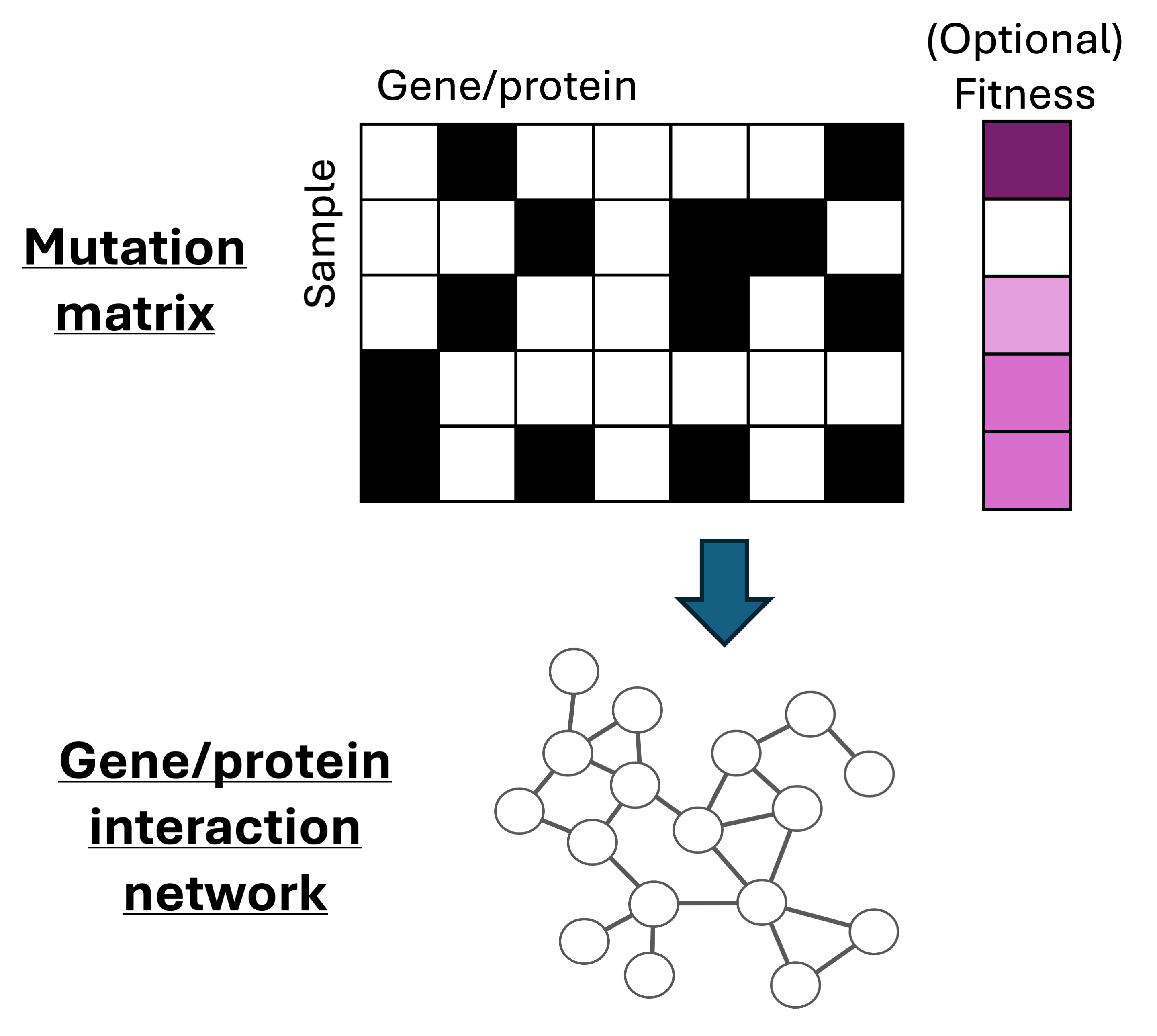
Biological networks underlie many aspects of human health and disease, but existing networks are highly incomplete (missing ≈90% of edges). We develop statistical methodologies for inferring biological interaction networks from noisy data (e.g. genetic perturbations or mutation). We have also developed theoretical approaches for analyzing networks in biology and other disciplines. Key words: graphs/networks, hypergraphs, epistasis, multivariate Bernoulli, genetic perturbations
|
|
Anomaly detection
|
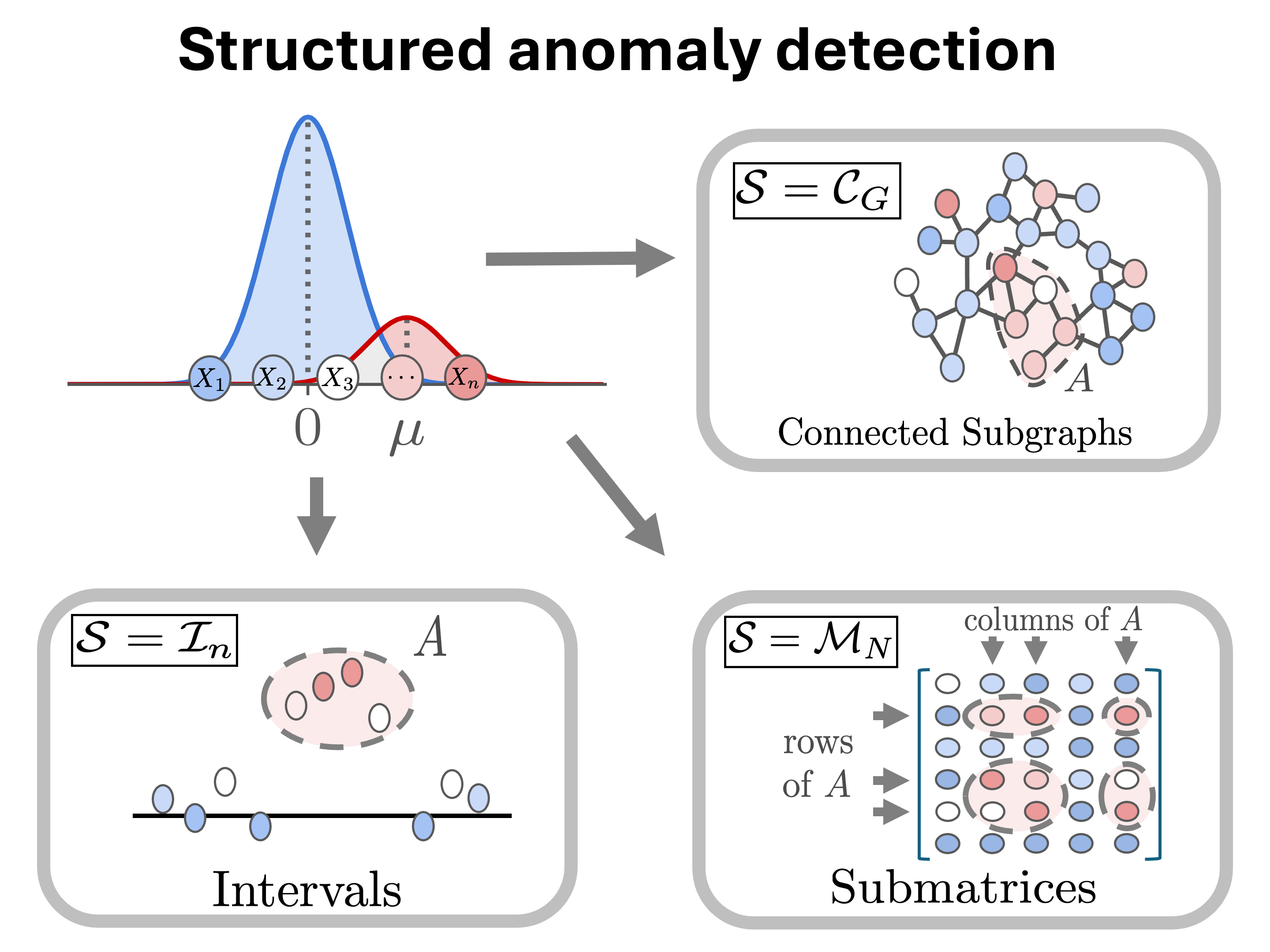
Anomalous interactions between genes/proteins underlie many complex diseases. We develop methods for anomaly detection in networks and other structured data (e.g. time series, matrices). Key words: protein-protein interaction (PPI) network, network anomaly detection, scan statistics, statistical bias
|
|